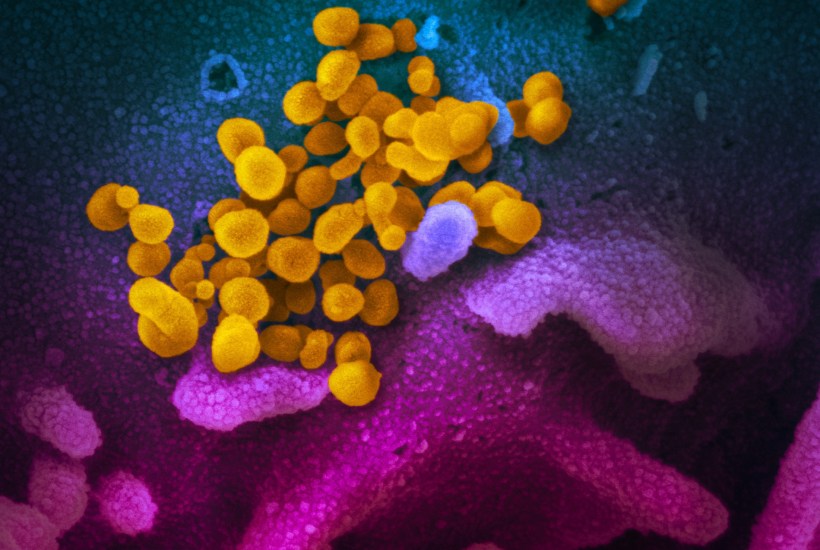
Common wisdom states that a new pathogen, once introduced into a vulnerable human population with no immune defences, will evolve over time to grow more benign and live in amity with its host. After all, the argument goes, a pathogen will depend on its host for its own survival, so causing mass disease and death will make onward transmission more difficult and ultimately only ensure its own demise.
Already a subscriber? Log in
Get 10 issues
for $10
Subscribe to The Spectator Australia today for the next 10 magazine issues, plus full online access, for just $10.
- Delivery of the weekly magazine
- Unlimited access to spectator.com.au and app
- Spectator podcasts and newsletters
- Full access to spectator.co.uk
Or


















Comments
Don't miss out
Join the conversation with other Spectator Australia readers. Subscribe to leave a comment.
SUBSCRIBEAlready a subscriber? Log in